Sindrome dell'occhio di gatto
🐱 La conoscevi? La sindrome dell’occhio di gatto è una malattia genetica molto rara causata da un'anomalia del cromosoma 22 ed è così chiamata per le malformazioni a carico di viso ed occhi presenti in alcuni pazienti. Scopriamola insieme!

La sindrome dell’occhio di gatto (Cat Eye Syndrome, CES) è una malattia genetica molto rara, con un’incidenza di 1:150.000 nati vivi, i cui segni e sintomi caratteristici sono dovuti alla presenza nelle cellule di copie extra del cromosoma 22, in particolare della regione 22q11.2. Tale sindrome è nota con altri nomi quali: tetrasomia parziale del cromosoma 22, cromosoma 22 duplicato invertito e sindrome di Schmid-Fraccaro e la trisomia parziale del cromosoma 22.
Gli individui affetti da CES possono presentare un’ampia gamma di disturbi che colpiscono diverse regioni dell’organismo, con particolare frequenza per la triade costituita dal coloboma oculare, l'atresia anale e le etichette/fossette cutanee preauricolari. Nella ricerca delle modalità di eredità del disturbo si è notato che difficilmente e sporadicamente questo si manifesta in individui che non presentano CES nella propria storia familiare.
Descrizione clinica
Haab e Schachenmann furono i primi a proporre il termine “sindrome dell’occhio di gatto”, in seguito ad osservazioni delle caratteristiche fenotipiche tipiche dei pazienti. La CES rappresenta una patologia cromosomica con uno spettro clinico vasto sia per caratteristiche che per gravità delle stesse, infatti, i soggetti affetti possono avere un fenotipo che varia nell’intervallo che va da lieve a grave malattia multi-sistemica, anche tra membri della stessa famiglia. Infatti, esistono diverse malformazioni, anche rare, associate a questa sindrome ma tre in particolare vanno a costituire la triade classica di sintomi associati alla CES, pur essendone stata stimata la presenza solo nel 41% di pazienti:
1. COLOBOMA OCULARE (Figura 1): il coloboma è un difetto dell’occhio che consistenella parziale assenza di tessuto oculare e che spesso interessa entrambi gli occhi (bilaterale). Questo difetto può colpire l’iride, la retina e/o coroide. Solitamente nella CES è presente in poco più della metà degli individui ed interessa esclusivamente l’iride, andando in questo modo a darne una forma irregolare che appare come una lacuna subito sotto la pupilla. Di conseguenza, la pupilla allungata ricorderà l’aspetto tipico dell’occhio di ungatto, da cui il nome della sindrome. Se il coloboma colpisce solo l’iride non ci sarà interruzione del campo visivo; mentre se dovesse colpire la retina l’area interessata sarà “cieca”.
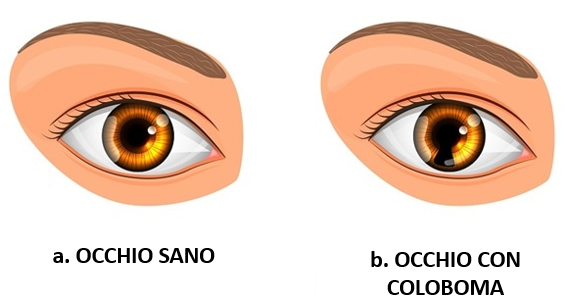
1. ATRESIA ANALE CON FISTOLA: si tratta di un disturbo in cui il canale anale risulta stretto o assente, con la presenza di una fistola. Nei maschi la fistola ha probabilità di formazione tra retto e vescica, uretra o perineo; mentre nelle femmine le fistole possono interessare la zona tra retto e la vescica o la vagina. Atresia e fistole anali vengono corrette chirurgicamente.
2. ETICHETTE CUTANEE E/O FOSSETTE PREAURICULARI: è l’anomalia più frequente negli individui affetti da CES, presente in più dell’80% dei casi. Le etichette sono piccole escrescenze della pelle, mentre le fossette sono depressioni lievi localizzate davanti alle orecchie esterne (preauricolari). I padiglioni auricolari possono essere bassi e/o malformati, ed in alcuni casi i canali dell’orecchio esterno risultano essere cechi o assenti (microtia). Nella maggior parte dei casi si verifica l’assenza del condotto uditivo esterno causando una lieve compromissione dell’udito poiché si ha una trasmissione inadeguata del suono dall’orecchio esterno a quello interno.
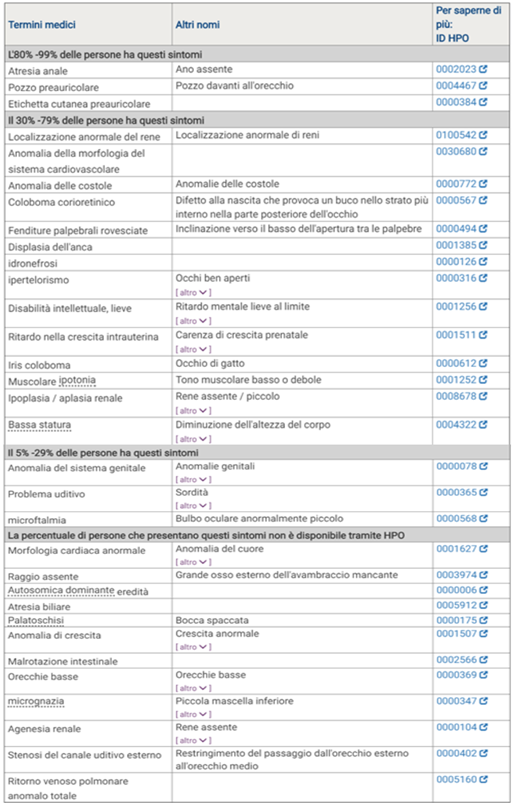
Altri sintomi associati alla sindrome (Tabella 1) sono legati non solo ad anomalie dell’occhio, anali e dell’orecchio ma anche dovuti a difetti cardiaci, difetti renali e genitali, ritardo mentale, difetti scheletrici, difetti addominali, bassa statura, palatoschisi, caratteristiche facciali anormali:
a. DIFETTI CARDIACI: alla nascita metà dei pazienti affetti da CES presenta difetti cardiaci congeniti come il "ritorno venoso polmonare anomalo totale" (TAPVR) o "tetralogia di Fallot" (TOF).
b. DIFETTI UROGENITALI: i difetti renali tipici associati al CES comprendono il sottosviluppo di uno o entrambi i reni (ipoplasia renale unilaterale o bilaterale), assenza di un rene (agenesi unilaterale), la presenza di un rene aggiuntivo (rene soprannumerario) etc. Mentre i difetti legati al tratto riproduttivo nelle femmine portano al sottosviluppo dell’utero, assenza della vagina o genitali anomali; nei maschi criptorchidismo e anomalie genitali esterne.
c. RITARDO MENTALE: alcuni dei pazienti presentano un normale sviluppo cognitivo, tuttavia la maggior parte di essi manifesta disabilità intellettuale che varia tra lieve, moderata e grave. Gli individui con disabilità intellettiva possono sperimentare ritardi nel raggiungimento di traguardi dello sviluppo che richiedono il coordinamento dell'attività muscolare e mentale (ritardi psicomotori).
d. DIFETTI SCHELETRICI: tipici difetti scheletrici possono essere scoliosi, fusioni vertebrali, assenza o fusione anomala di alcune costole, assenza di alcune dita dei piedi e/o duplicazioni degli alluci.
e. DIFETTI ADDOMINALI: comprendono ad esempio ernia ombelicale o ernia inguinale, ovvero parti dell’intestino che sporgono rispettivamente attraverso un difetto della parete addominale dell’ombelico o nel canale che attraversa gli strati muscolari inferiori della parete addominale; malattia di Hirschsprung, che comporta la rotazione incompleta dell'intestino crasso superiore (cieco) e / o l'assenza di gruppi di fibre nervose (gangli) nella parete muscolare dell'intestino crasso con conseguente compromissione o assenza delle contrazioni ritmiche involontarie (peristalsi) che spingono i materiali di scarto attraverso il tratto digestivo inferiore (possono includere un accumulo anormale di feci all'interno del colon); infine atresia biliare.
f. PALATOSCHISI: si tratta di malformazione del palato, che si presenta come una fenditura più o meno estesa della parte anteriore del palato duro. Qualora la fenditura prosegua coinvolgendo anche il labbro superiore (cheiloschisi) si parla di cheilognatopalatoschisi o labiopalatoschisi. Nei pazienti affetti da CES questo difetto si osserva nel 14-31% dei casi con gradi diversi.
g. BASSA STATURA: riscontrata nel 15-50% dei casi di CES e potrebbe essere associata ad una carenza di ormone della crescita nei pazienti affetti, tuttavia, non è ancora del tutto chiara questa teoria.
h. CARATTERISTICHE FACCIALI ANORMALI: i pazienti affetti da CES tendono a presentare caratteristiche anomalie della regione craniofacciale che comprendono occhi ampiamente distanziati, ponte nasale piatto, mascella inferiore anormalmente piccola, le pieghe della palpebra inclinate verso il basso (ragadi palpebrali) e pieghe della pelle verticali che possono coprire gli angoli interni degli occhi (pieghe epicantali).
Tutti questi sintomi sono causati da uno sviluppo anomalo sia nel periodo embrionale che fetale e, anche se l’aspettativa di vita non è significativamente ridotta, alcuni pazienti muoiono a causa di più malformazioni durante la prima infanzia.
Cromosoma 22
La CES è provocata da un’anomalia cromosomica che interessa il cromosoma 22. I ricercatori che presero parte al Progetto Genoma Umano, nel 1999, sequenziarono completamente il cromosoma 22, cioè il primo cromosoma umano completamente sequenziato. Parliamo del secondo cromosoma più piccolo rappresentante 1,5-2% del DNA totale cellulare e che copre 51 milioni di coppie di basi. Questo cromosoma contiene probabilmente da 500 a 600 geni codificanti per proteine con funzioni variegate all’interno dell’organismo ed è, inoltre, soggetto a diversi cambiamenti relativi alla sua struttura o il numero di copie, che lo rendono principale responsabile di numerosi disturbi oltre la CES, come ad esempio la sindrome di DiGeorge, sindrome di Phelan-McDermid, leucemia mieloide cronica, dermatofibrosarcoma protuberans, la sindrome di Emanuel, sarcoma di Ewing, sindrome di Opitz G/BBB.
In particolare, i pazienti affetti da CES presentano un cromosoma 22 con un frammento invertito e duplicato (inv. dup. 22) (Figura 2). Di conseguenza, nelle cellule dei pazienti colpiti dalla sindrome dell’occhio di gatto si riscontra un cromosoma 22 extra, ovvero un cromosoma marcatore bisatellito soprannumerario, che è alla base dei segni e sintomi caratteristici della sindrome: in caso di CES si riscontra la presenza di quattro copie del cromosoma 22 (tetrasomia parziale), due volte come parte dei due cromosomi normali 22 e due volte insieme nel cromosoma marcatore. Inoltre, in alcune persone, questo cromosoma extra può essere presente solo in una determinata percentuale delle cellule del corpo (mosaicismo).

Il locus genetico della Cat Eye Syndrome è conosciuto come CESCR (Cat Eye Syndrome Critical Region), e corrisponde alla regione cromosomica 22q11.2 indicata anche come 22pterq11 (Fig.11). Tuttavia, la CES non è l’unica patologia associata al CESCR. Un esempio è rappresentato dalla Sindrome di DiGeorge, che interessa la medesima regione cromosomica della Sindrome dell’occhio di gatto.
Molti disturbi genomici che comportano delezioni o duplicazioni di almeno 1 Mb (1 milione di coppie di basi) sono associati ai blocchi di ripetizioni a bassa copia (LCR) specifici, e si ritiene che questi LCR stimolino eventi di ricombinazione omologa meiotica. La regione del cromosoma 22q11.2 è suscettibile ai riarrangiamenti, mediati da ripetizioni a bassa copia (LCR22). Quindi, Le delezioni e le duplicazioni che interessano tale regione sono mediate da eventi di ricombinazione omologa tra LCR22, che nel caso della CES sono responsabili della formazione del cromosoma extra inv. dup. 22.Il meccanismo preciso per i diversi riarrangiamenti mediati da LCR22 non è ancora determinato; tuttavia, è chiaro che questi sono implicati nella genesi del disturbo e quindi hanno valore clinico significativo.
Ereditarietà
L'anomalia cromosomica caratteristica della CES molto frequente ha insorgenza “de novo”, mentre raramente viene trasmessa da genitore a figlio e sporadicamente è dovuta ad errori meiotici. Quindi, il cromosoma marcatore può essere ereditato sia da genitori sani, per errori nella divisione dei gameti, che da genitori affetti da CES, che trasmettono il frammento cromosomico extra alla prole.
Inoltre, in letteratura si possono trovare segnalazioni di casi in cui il mosaicismo per l’anomalia cromosomica viene trasmesso attraverso diverse generazioni nelle famiglie, i cui componenti hanno alta variabilità nell’espressioni dei sintomi che comprende casi lievi ma anche gravi. Non ci sono dati relativi al rischio di ricorrenza tra fratelli e sorelle di un paziente affetto da CES, tuttavia, dopo la nascita di un bambino affetto è necessario un esame cromosomico di entrambi i genitori.
Inoltre, se l’esame cromosomico a livello dei linfociti indica un cariotipo normale non si può escludere un mosaicismo nascosto che può interessare la linea germinale e comporta un piccolo rischio di ricorrenza. Per la prole di un paziente fertile affetto da CES il rischio di insorgenza sarà vicino al 50%.
Metodi diagnostici
In presenza di manifestazioni cliniche caratteristiche, la diagnosi di CES si avvale della ricerca di materiale cromosomico extra derivato dal cromosoma 22, che può essere rilevato tramite test citogenetici quali ad esempio l’analisi del cariotipo e la FISH (utile nel rilevamento di un mosaicismo di basso livello). La sindrome può essere diagnosticata sia in fase prenatale che in fase postnatale, affiancate sempre da una diagnosi differenziale:
1. DIAGNOSI PRENATALE (prima del parto): nei casi in cui l’ecografia fetale evidenzi potenziali difetti che facciano sospettare la sindrome dell’occhio di gatto, verranno eseguiti ulteriori test per giungere ad una diagnosi effettiva:
1.1 ULTRASUONI: questo test consente di individuare malformazioni e anomalie fetali più importanti fin dal primo trimestre. Solitamente si effettua un primo screening con gli ultrasuoni tra l’ 11a e la 14asettimana per individuare anomalie più evidenti, ed un secondo screening tra la 18a e la 20a settimana per evidenziare le anomalie strutturali minori (https://www.testprenataleaurora.it/it/blog/13-test-prenatali/899-il-ruolo-degli-ultrasuoni-nella-diagnosi-prenatale.html).
1.2 AMNIOCENTESI: è una tecnica invasiva utilizzata nella diagnosi prenatale che consiste nel prelievo di liquido amniotico dalla cavità uterina, solitamente effettuata tra la 16a e 18a settimana. Il liquido amniotico contiene cellule fetali in sospensione (amniociti) di cui verrà analizzato e studiato l’assetto cromosomico. Quindi attraverso questo test si ottiene l’analisi del cariotipo fetale per poter evidenziare l’eventuale presenza di anomalie e diagnosticare eventuali disturbi genetici (http://www.diagnosiprenatale.com/diagnosi-prenatale/amniocentesi-scheda-tecnica.aspx).
1.3 PRELIEVO DEI VILLI CORIALI (chorionic villus sampling, CVS): tecnica invasiva eseguita tra la 10a e 12asettimana, che prevede il prelievo da una regione della placenta di un campione di tessuto trofoblastico. Sul campione prelevato tramite CVS si possono effettuare indagini citogenetiche, indagini biochimiche ed analisi del DNA (è la metodica d’elezione per questo tipo di indagine) (https://www.saperidoc.it/flex/cm/pages/ServeBLOB.php/L/IT/IDPagina/102)
2. DIAGNOSI POSTNATALE (dopo il parto): ha lo scopo di accertare la presenza di possibili anomalie cromosomiche che possono essere associate ad una condizione di patologia genetica, ad una riduzione della fertilità o ad una maggiore probabilità di generare figli affetti da una patologia genetica. L’indagine viene approfondita, ed eventualmente confermata la diagnosi, attraverso studi cromosomici standard.
L’esecuzione dell’analisi del cariotipo in periodo postnatale si effettua in casi di: sospetta sindrome cromosomica, malformazioni congenite, disordini neurologici, autismo, epilessia, disordini cognitivi e dello sviluppo psicomotorio, approfondimento diagnostico per la caratterizzazione di anomalie cromosomiche precedentemente individuate attraverso il cariotipo convenzionale, fenotipi complessi e familiari di soggetti con anomalie cromosomiche (https://www.microgenomics.it/servizi/diagnosi-postnatale/#:~:text=Diagnosi%20postnatale%20%E2%80%93%20Microgenomics-,Diagnosi%20postnatale,affetti%20da%20una%20patologia%20genetica.).
La diagnosi postnatale si avvale di un’approfondita valutazione clinica per la rivelazione di più difetti fisici caratteristici della patologia sospettata, nel caso di CES coloboma oculare, atresia anale, etichette e/o fossette auricolari.
3. DIAGNOSI DIFFERENZIALE: con la diagnosi differenziale si va ad escludere, in un gruppo di patologie con sintomi e segni simili, la condizione o patologia effettiva del paziente (https://medicinaonline.co/2018/03/22/differenza-tra-diagnosi-e-diagnosi-differenziale/).
I disturbi cromosomici con fenotipi sovrapposti alla CES sono la Sindrome di CHARGE e l’Associazione VECTERL, che sono state precedentemente trattate. Per effettuare diagnosi differenziale nel caso della CES possono essere eseguiti diversi test per evidenziare l’eventuale presenza di caratteristiche secondarie tipiche come, ad esempio, una valutazione cardiaca approfondita per la rivelazione di anomalie cardiache; attenta visita oculistica; monitoraggio dell’udito; tecniche di imaging e/o altri test per possibili difetti gastrointestinali, renali, genitourinari, scheletrici o biliari; studio della funzione cognitiva.
Gestione e trattamento
Per il trattamento della sindrome dell’occhio di gatto è necessaria l’organizzazione di un team di professionisti in diversi campi medici quali pediatri, chirurgi, cardiologi, ortopedici, oculisti, gastroenterologhi e ancora altri operatori sanitari. Ci si avvale per questa sindrome di una gestione multidisciplinare che dipenderà dalla gravità e dai segni e sintomi caratteristici presentati da ogni paziente, in modo da poter intervenire in modo specifico verso le diverse anomalie associate alla sindrome attraverso l’uso di farmaci, intervento chirurgico e/o altre misure.
Quando la terapia comprende l’intervento chirurgico, sia prima che dopo gli interventi, i pazienti possono essere soggetti ad infezioni batteriche che, quindi, devono essere anticipate e trattate vigorosamente con terapia antibiotica preventiva.
Molto importante per il trattamento di questa sindrome è effettuare un intervento precoce e a tal fine risultano utili i servizi speciali comprendenti l’educazione correttiva, il supporto sociale speciale e/o ulteriori servizi di natura medica, sociale e professionale. Inoltre, si raccomanda l’indagine cromosomica sia ai genitori di soggetti affetti da CES che a soggetti adulti affetti dalla sindrome interessati ad avere figli.
Prognosi
La prognosi per individui affetti da CES è variabile tra gli individui e dipende dalla gravità della condizione e dai segni e sintomi ad essa associati, soprattutto in presenza di difetti cardiaci o renali. La maggior parte dei pazienti non ha aspettativa di vita ridotta in modo significativo, tuttavia si registrano alcuni casi in cui si verifica il decesso dei pazienti nella prima infanzia a causa di gravi malformazioni.
Lidia Caserta
Fonti
- Berends, M.J., Tan-Sindhunata, G., Leegte, B., van Essen, A.J, 2001. Phenotypic variability of Cat-Eye syndrome. Genet Couns. 12, 23-34. https://pubmed.ncbi.nlm.nih.gov/11332976/;
- Brewer, C., Holloway, S., Zawalnyski, P., Schinzel, A., FitzPatrick, D., 1998. A Chromosomal Deletion Map of Human Malformations. The American Journal of Human Genetics 63, 1153–1159. https://doi.org/10.1086/302041;
- Chen, H. (Ed.), 2012. Cat Eye Syndrome, in: Atlas of Genetic Diagnosis and Counseling. Springer US, New York, NY, 279–283. https://doi.org/10.1007/978-1-4614-1037-9_33;
- Edelmann, L., 1999. A common molecular basis for rearrangement disorders on chromosome 22q11. Human Molecular Genetics 8, 1157–1167. https://doi.org/10.1093/hmg/8.7.115;
- Lüleci, G., Bağci, G., Kivran, M., Lüleci, E., Bektaş, S., Başaran, S., 1989. A hereditary bisatellite-dicentric supernumerary chromosome in a case of cat-eye syndrome. Hereditas 111, 7-10. https://doi.org/10.1111/j.1601-5223.1989.tb00369.x;
- Pierson, M., Gilgenkrantz, S., Saborio, M., 1975. Syndrome dit de l'oeil de chat avec nanisme hypophysaire et developpement mental normal. Arch. Franc. Pediatr. 32, 835-848;
- Quintero-Rivera, F., Martinez-Agosto, J.A., 2013. Hemifacial microsomia in cat-eye syndrome: 22q11.1-q11.21 as candidate loci for facial symmetry. Am. J. Med. Genet. 161, 1985–1991. https://doi.org/10.1002/ajmg.a.35895;
- Rosias, P.R., Sijstermans, J.M., Theunissen, P.M., Pulles-Heintzberger, C.F., De Die-Smulders, C.E., Engelen, J.J., Van Der Meer, S.B., 2001. Phenotypic variability of the cat eye syndrome. Case report and review of the literature. Genet Couns. 12, 273-282. https://pubmed.ncbi.nlm.nih.gov/11693792/;
- Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1116/;
- https://www.news-medical.net/health/What-is-Coloboma.aspx;
- https://rarediseases.info.nih.gov/diseases/26/disease;
- https://www.testprenataleaurora.it/it/blog/13-test-prenatali/899-il-ruolo-degli-ultrasuoni-nella-diagnosi-prenatale.html;
- http://www.diagnosiprenatale.com/diagnosi-prenatale/amniocentesi-scheda-tecnica.aspx;
- https://www.saperidoc.it/flex/cm/pages/ServeBLOB.php/L/IT/IDPagina/102;
- https://www.microgenomics.it/servizi/diagnosi-postnatale/#:~:text=Diagnosi%20postnatale%20%E2%80%93%20Microgenomics-,Diagnosi%20postnatale,affetti%20da%20una%20patologia%20genetica.;
- https://medicinaonline.co/2018/03/22/differenza-tra-diagnosi-e-diagnosi-differenziale/;
- https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg38&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chr22%3A8101986%2D18601986&hgsid=865318969_nM03puAkFVhwsW13XXNrZAA5aWeV.
Ti è piaciuto l'articolo?
 BioDaily.it non riceve alcun contributo pubblico né ospita alcuna pubblicità, quindi si sostiene esclusivamente grazie alle donazioni dei lettori. Ti ringraziamo qualora tu volessi fare una donazione al nostro progetto, puoi farlo cliccando su questo messaggio.
BioDaily.it non riceve alcun contributo pubblico né ospita alcuna pubblicità, quindi si sostiene esclusivamente grazie alle donazioni dei lettori. Ti ringraziamo qualora tu volessi fare una donazione al nostro progetto, puoi farlo cliccando su questo messaggio.