Distrofia muscolare di Duchenne
👩🏻⚕️ La distrofia muscolare di Duchenne (DMD) è una patologia X-linked. Nel gruppo delle distrofie muscolari rappresenta il disturbo maggiormente frequente, con degenerazione progressiva e inarrestabile delle fibre muscolari. Scopriamola insieme!
La distrofia muscolare di Duchenne (DMD; OMIM 300377) è una patologia X-linked con incidenza abbastanza elevata, mediamente 1/3.500 maschi nati vivi. In particolare, nel gruppo delle distrofie muscolari, la DMD rappresenta il disturbo muscolare maggiormente frequente con degenerazione progressiva e inarrestabile delle fibre muscolari.
L’esordio della patologia si ha nei primi mesi di vita ed è molto difficile capire quando inizia realmente la malattia in quanto il danno alle membrane inizia già con i movimenti del feto nell’utero; tuttavia, al momento della nascita i bambini hanno una qualità di massa muscolare buona per consentire la diagnosi.
DISTROFIE MUSCOLARI
Le distrofie muscolari sono patologie in cui si vede il coinvolgimento della muscolatura striata, in modo particolare di molecole di connessione fondamentali per il corretto assemblaggio delle fibre contrattili ed il loro scorrimento.
Quindi, in caso di alterazione di queste molecole si ha un’alterazione dell’intera funzione muscolare, in quanto non si riesce a trasferire il movimento al sarcolemma. In questo caso, si genera danno alle membrane che sfocia in morte cellulare precoce, con conseguente infiammazione e fibrosi.
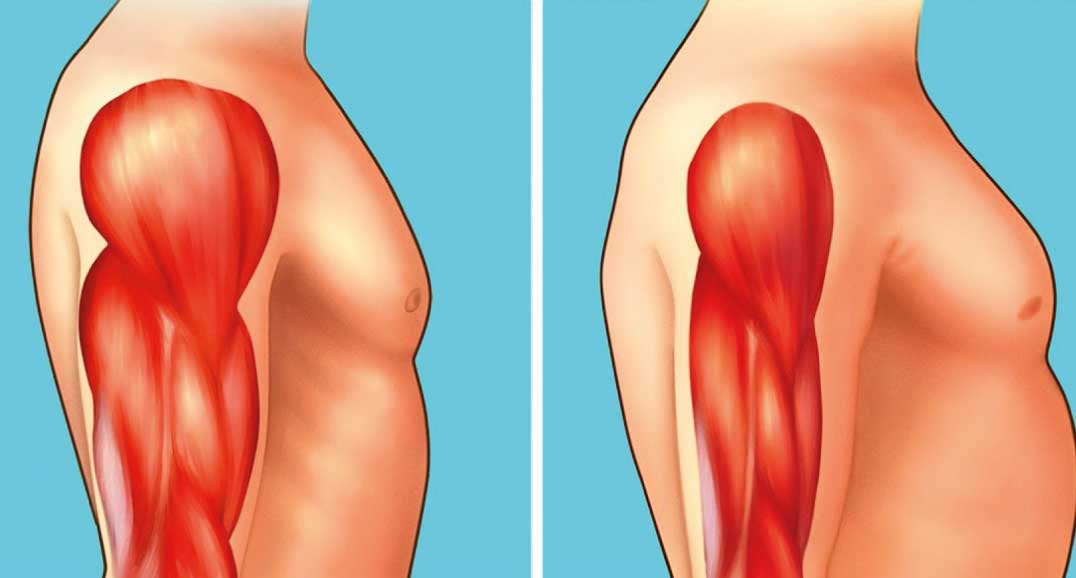
Inoltre, la riserva staminale non riesce più a sopperire a questa degenerazione cellulare e nelle zone precedentemente occupate da cellule si generano cicatrici fibro-connettivali. Tutto questo porta alla formazione di un muscolo distrofico (Figura 1), da cui la definizione di questo gruppo di malattie associate da alterazioni simili ma causate da mutazioni che colpiscono geni differenti.
GENE & PROTEINA
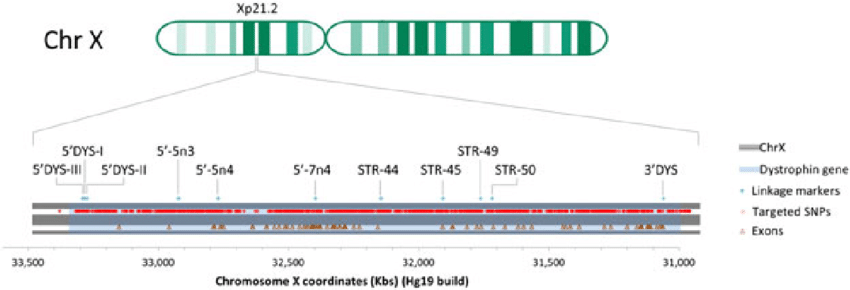
Il gene causativo della distrofia muscolare di Duchenne è il gene della distrofina, localizzato sul cromosoma X (Xp21.2) (Figura 2). Nel caso della DMD, si possono verificare mutazioni di diverso tipo lungo tutto il gene che portano alla sintesi di una proteina non funzionale. Infatti, le delezioni rappresentano l’80% delle mutazioni coinvolte nella patologia, mentre il 20% comprende duplicazioni, microinserzioni, microdelezioni e mutazioni puntiformi.
Essendo una patologia X-linked sono prevalentemente colpiti sono i maschi, che ricevono il cromosoma X con l’allele mutato dalla madre portatrice. Le donne, invece, sono solitamente portatrici ma ci sono rari casi in cui anche nelle donne si riscontra la sintomatologia tipica della malattia, questo si verifica in donne omozigoti per disomia uniparentale, entrambi i cromosomi sono ereditati dalla madre per non disgniunzione oppure nel caso in si ha genotipo X0.
Ad esempio, nelle donne normalmente entrambi i cromosomi X sono compattati al 50% nei diversi citotipi cellulari, tuttavia, può accadere che uno dei due sia ipercompattato, cioè compattato in quasi tutte le cellule dell’organismo e se il cromosoma residuo contiene l’allele malato la patologia si manifesta.
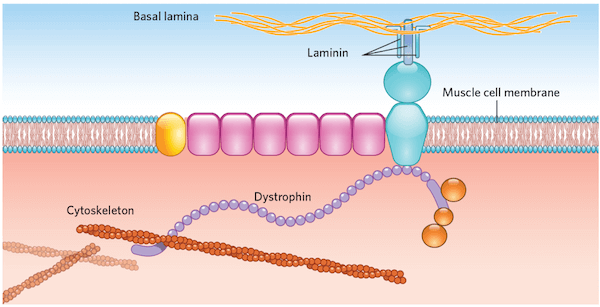
La proteina coinvolta nella DMD è la distrofina, ovvero una proteina che connette i filamenti di actina con strutture di membrana quali sarcoglicani, consentendo l’interazione con il sarcolemma. Quindi, alterazioni nella sintesi di questa proteina si traducono con l’esordio della patologia.
In particolare, la distrofina è costituita da due domini globulari localizzati alle due estremità, N-terminale e C- terminale, che permettono l’interazione con l’actina e con le strutture di ancoraggio della membrana.
Nella distrofia muscolare di Duchenne le mutazioni portano alla perdita completa della proteina oppure la sintesi di una proteina anomala con un solo dominio, ed in questo caso la distrofina non sarà in grado di collegare le strutture contrattili con il sarcolemma (Figura 3).
Il danno principale, quindi, si verifica a livello del sarcolemma a livello del quale si verifica una sostituzione del tessuto muscolare con fibre connettivali e tralci adiposi, con conseguente riduzione della capacità contrattile diminuisce. Quindi ci sarà degenerazione del tessuto muscolare, con continua lacerazione delle membrane, dovuta al disaccoppiamento tra le fibre di actina e miosina con il sarcolemma.
SINTOMI
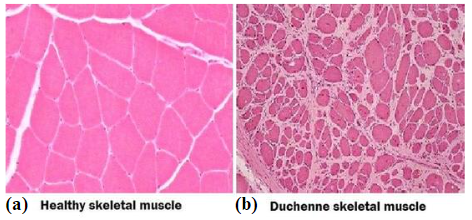
Come detto in precedenza, è complesso capire quando inizia realmente la malattia nonostante i danni alle membrane generati in periodo fetale. Tuttavia, questi danni alle membrane portano alla manifestazione del fenomeno tipico delle distrofie muscolari, e quindi anche nella DMD, quale la sostituzione del tessuto necrotico (tessuto muscolare) con una cicatrice fibro-connettivale o fibro-adiposa (tessuto fibroso e adiposo) (Figura 4).
Di conseguenza, da un punto di vista istopatologico le distrofie sono caratterizzata dalla presenza di:
1. Fibre disomogenee;
2. Aree di necrosi;
3. Presenza di infiltrato fibro-connettivale o fibro-adiposo.
Nella distrofia muscolare di Duchenne, i sintomi veri e propri si iniziano notare quando la capacità di recupero da parte della riserva staminale comincia a venir meno, ovvero tra i 2 e i 5 anni, con particolare evidenza a livello del cingolo pelvico e cingolo scapolare.
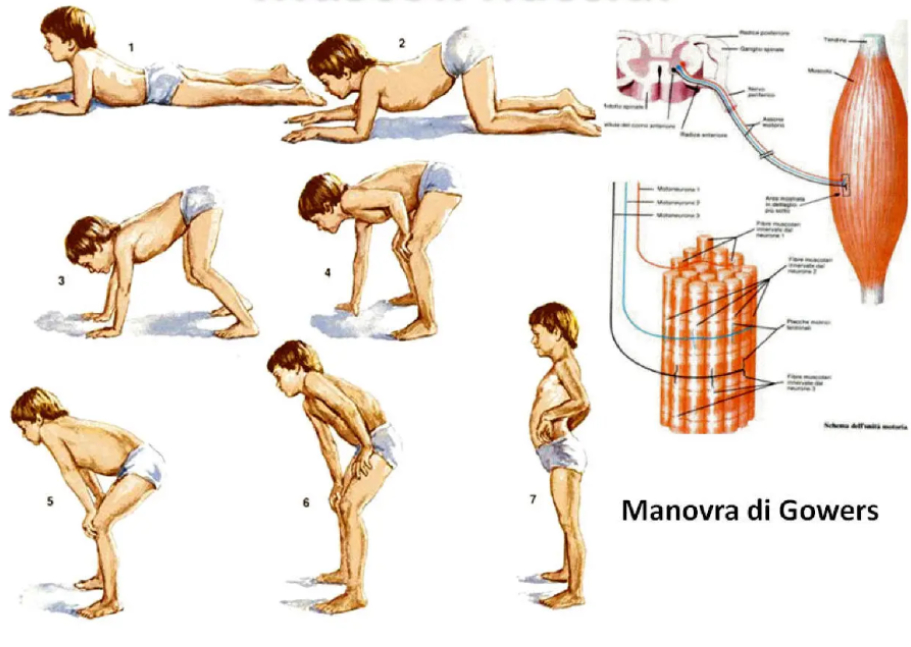
Inoltre, la DMD si distingue dalle altre distrofie per un particolare sintomo caratteristico quale è la difficoltà che hanno i bambini nell'alzarsi da terra. Infatti, i bambini affetti da DMD hanno una debolezza intrinseca dei muscoli dei glutei e delle cosce, per cui nell'alzarsi da terra effettuano un movimento noto come “tecnica dell'arrampicata” o manovra di Gowers, durante il quale si aiutano aggrappandosi alle proprie gambe e alle ginocchia per tirarsi in piedi (Figura 5).
Inoltre, crescendo anche i muscoli respiratori ed il cuore iniziano ad essere coinvolti sfociando in una sindrome disventilatoria restrittiva, cioè dovuta alla mancata espansione della gabbia toracica.
Questo può portare alla necessità della ventilazione meccanica in un primo momento, che sarà soltanto notturna all’inizio, fino all’utilizzo del polmone d’acciaio, ovvero una macchina che consente l’espansione della gabbia toracica. In particolare, il coinvolgimento cardiaco porta a miocardiopatia dilatativa si evidenzia soprattutto in quei pazienti che arrivano ad una sopravvivenza prolungata.
Infatti, oggi che possono essere messi nei polmoni di acciaio e che la sopravvivenza si è allungata, questi pazienti muoiono per insufficienza cardiocircolatoria e cardiomiopatia dilatativa. L’aspettativa di vita, con questi segni clinici, è di circa 15/20 anni.
Tra gli altri sintomi della patologia abbiamo:
1. Difficoltà nei movimenti: correre, salire le scale, alzarsi da terra, saltare.
2. Pseudo-ipertrofia del polpaccio: sembra che i pazienti abbiano dei polpacci molto sviluppati, ma in realtà il muscolo, che è sottoposto ad un ingente carico per la tipica “camminata” dei bambini in punta dei piedi, si è ricco di tessuto fibro-connettivale e tessuto adiposo.
3. Iper-lordosi lombare e scapole alate a causa della debolezza dei muscoli del cingolo scapolare.
4. Camminata nota come “andatura del maratoneta”: i bambini iniziano a camminare poggiando il tallone piuttosto che la punta dei piedi sulla superficie con movimento dell'anca caratteristico.
5. Complicanze cardiache e respiratorie: intorno ai 12 anni di età insorgono: difficoltà di movimento degli arti superiori.
DIAGNOSI
Quando si sospetta che un bambino possa essere affetto da questa DMD, inizialmente il medico richiede il dosaggio della CPK (fosfo-creatin chinasi) che nei pazienti affetti è 10 volte superiore della norma, mentre nelle donne portatrici è nel 20-30% dei casi di 2-3 volte aumentata.
Poiché si tratta di una patologia provocata nella maggior parte dei casi da delezioni, un altro metodo per la diagnosi è la ricerca lungo la sequenza genica di queste mutazioni con MLPA e MULTIPLEX PCR.
Tuttavia, se non dovessero risultare delezioni o duplicazioni, si progsegue con la ricerca di mutazioni meno frequenti (mutazioni non senso, silenti, microinserzioni e microdelezioni) mediante tecniche di pre-screening (DDGE, SSCP, DHPLC) o attraverso il sequenziamento.
Lidia Caserta
Fonti
1. Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1116/
2. M. Parks, S. Court, S. Cleary, S. Clokie, J. Hewitt, D. Williams, T. Cole, F. MacDonald, M. Griffiths and S. Allen. (2016). Non-invasive prenatal diagnosis of Duchenne and Becker muscular dystrophies by relative haplotype dosage. Prenatal diagnosis, 36, 312–320. DOI: 10.1002/pd.4781
3. Zatz M, Rapaport D, Vainzof M, Passos-Bueno MR, Bortolini ER, Pavanello R de C, Peres CA. Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. J Neurol Sci. 1991;102:190–6.
4. Salmaninejad A, Valilou SF, Bayat H, Ebadi N, Daraei A, Yousefi M, Nesaei A, Mojarrad M. Duchenne muscular dystrophy: an updated review of common available therapies. Int J Neurosci. 2018 Sep;128(9):854-864
5. Zalaudek I, Bonelli RM, Koltringer P, Reisecker F, Wagner K. Early diagnosis in Duchenne muscular dystrophy. Lancet. 1999;353:1975.
6. White S, Kalf M, Liu Q, Villerius M, Engelsma D, Kriek M, Vollebregt E, Bakker B, van Ommen GJ, Breuning MH, den Dunnen JT. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet. 2002;71:365–74.
7. Viollet L, Thrush PT, Flanigan KM, Mendell JR, Allen HD. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am J Cardiol. 2012;110:98–102.
8. Viggiano E, Ergoli M, Picillo E, Politano L. Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. Hum Genet. 2016;135:685–98.
Ti è piaciuto l'articolo?
 BioDaily.it non riceve alcun contributo pubblico né ospita alcuna pubblicità, quindi si sostiene esclusivamente grazie alle donazioni dei lettori. Ti ringraziamo qualora tu volessi fare una donazione al nostro progetto, puoi farlo cliccando su questo messaggio.
BioDaily.it non riceve alcun contributo pubblico né ospita alcuna pubblicità, quindi si sostiene esclusivamente grazie alle donazioni dei lettori. Ti ringraziamo qualora tu volessi fare una donazione al nostro progetto, puoi farlo cliccando su questo messaggio.